PipelineSimple: Advanced features¶
There are several extra features that you can attach to the PipelineSimple. In this section we’ll explain the database interface, checkpointing of experiments, multitasking with Dask, and score file formats.
The database interface¶
A database interface is responsible for providing data samples to the pipeline when requested so. It is the starting point of each sub-pipelines of PipelineSimple: Train, Enroll and Score.
Note
Since the bob packages do not include the actual data, the database interfaces need to be set up so that a path to the data is configurable. The interface only knows the file structure from that path and loads the data at runtime. Consult the documentation of each database to learn how to do this.
Groups of subjects are defined for the different parts of a biometric experiment.
The world set is used to train the model.
The dev set is used to enroll and score the subjects to define a working point for the system (setting a threshold value).
And for a complete experiment, the eval set is used to evaluate the performance of a biometric system on the working point given by the dev set scoring.
Each set must contain samples from individuals that are not represented in any other set.
Those set are then used in the experiment’s pipeline:
The world set is passed to the training sub-pipeline to fit the transformers’ background model.
Several samples (enough to enroll an individual) from each subject of the dev set are passed to the enroll sub-pipeline as references.
The rest of the samples of the dev set, the probes, are passed to the scoring sub-pipeline to generate the
scores-devfile.If needed, the last two steps are repeated for the eval set of subjects, to generate the
scores-evalfile.
Using a database interface¶
You may list the currently available databases (and pipelines) using the following command:
$ bob bio pipeline simple --help
You can use such a dataset with the following command (example with the AT&T dataset):
$ bob bio pipeline simple atnt <pipeline_name>
For more exotic datasets, you can simply pass your custom database file (defining a database object) to the PipelineSimple:
$ bob bio pipeline simple my_database.py <pipeline_name>
The database object defined in my_database.py is an instance of either:
A
bob.bio.base.database.CSVDataset(see here),A
bob.bio.base.database.CSVDatasetCrossValidation(see here),Your implementation of a Database Interface,
CSV file Database interface¶
The easiest way to create a database interface is to use the CSV file interface. This method is less complete and less flexible than implementing a full interface class but is sufficient for most use cases.
Protocol definition is possible and a set of CSV files (at least dev_enroll.csv and dev_probe.csv) in a folder must be created for each protocol.
The interface is created with bob.bio.base.database.CSVDataset.
This class takes as input the base directory and the protocol sub-directory of
the CSV file structure, and
finally, a csv_to_sample_loader that will load a sample data from a CSV row
read from the CSV files. This csv_to_sample_loader needs to know the dataset
base path and the extension of the dataset files.
File format¶
You must provide a series of comma separated values (CSV) files containing at least two columns with a header:
PATH,REFERENCE_ID
data/model11_session1_sample1,1
data/model11_session1_sample2,1
data/model11_session1_sample3,1
data/model11_session2_sample1,1
data/model12_session1_sample1,2
data/model12_session1_sample2,2
data/model12_session1_sample3,2
data/model12_session2_sample1,2
The required columns in each file are the path to a sample (header: PATH, relative to the dataset root) and a unique identifier for the individual represented by the sample (header: REFERENCE_ID).
Metadata¶
This interface allows metadata to be shipped with the samples. To do so, simply add columns in the CSV file, with a corresponding header:
PATH,REFERENCE_ID,MY_METADATA_1,METADATA_2
data/model11_session1_sample1,1,F,10
data/model11_session1_sample2,1,F,10
data/model11_session1_sample3,1,F,10
data/model11_session2_sample1,1,F,10
data/model12_session1_sample1,2,M,30
data/model12_session1_sample2,2,M,30
data/model12_session1_sample3,2,M,30
data/model12_session2_sample1,2,M,30
File structure¶
The following file structure and file naming must be followed, for the class to find the CSV files:
my_dataset
|
+-- my_protocol_1
| |
| +-- dev
| |
| +-- for_models
| +-- for_probes
|
+-- my_protocol_2
|
+-- norm
| |
| +-- train_world.csv
|
+-- dev
| |
| +-- for_models.csv
| +-- for_probes.csv
|
+-- eval
|
+-- for_models.csv
+-- for_probes.csv
The minimal required files are the
dev_enroll.csvanddev_probe.csv, containing the sample paths and subjects of the dev set.The
train.csvfile (as shown inmy_protocol_2) is optional and contains the information of the world set.The
eval_enroll.csvandeval_probe.csvfiles (as shown inmy_protocol_2) are optional and contain the information of the eval set.
In this example, my_dataset_csv_folder would be the base path given to the dataset_protocol_path parameter of bob.bio.base.database.CSVDataset, and my_protocol_1 the protocol parameter:
from bob.bio.base.database import CSVDataset, AnnotationsLoader
import bob.io.base
# Define a loading function called for each sample with its path
def my_load_function(full_path):
# Example with image samples
return bob.io.base.load(full_path)
# Create a loader that takes the root of the dataset and a loader function
my_sample_loader = CSVToSampleLoader(
data_loader=my_load_function,
dataset_original_directory="/path/to/dataset/root",
extension=".png",
metadata_loader=AnnotationsLoader()
)
# Create the CSV interface
database = CSVDataset(
name="my_dataset",
dataset_protocol_path="my_dataset_csv_folder",
protocol="my_protocol_1",
csv_to_sample_loader=my_sample_loader
)
This will create a database interface with:
The elements in
train_world.csvreturned bybob.bio.base.database.CSVDataset.background_model_samples(),The elements in
for_models.csvreturned bybob.bio.base.database.CSVDataset.references(),The elements in
for_probes.csvreturned bybob.bio.base.database.CSVDataset.probes().
An aggregation of all of the above is available with the
bob.bio.base.database.CSVDataset.all_samples() method, which returns
all the samples of the protocol.
CSV file Cross-validation Database interface¶
The bob.bio.base.database.CSVDatasetCrossValidation takes only one CSV file of identities and creates the necessary sets pseudo-randomly.
The format of the CSV file is the same as in bob.bio.base.database.CSVDataset, comma separated with a header:
PATH,REFERENCE_ID
path/to/sample0_subj0,0
path/to/sample1_subj0,0
path/to/sample2_subj0,0
path/to/sample0_subj1,1
path/to/sample1_subj1,1
path/to/sample2_subj1,1
Two set are created: a train set and a test set. By default, the ratio between these two sets is defined at 0.2 train subjects and 0.8 test subjects.
By default, one sample of each subject of the test set will be used for enrollment. The rest of the samples will be used as probes against the models created by the enrolled samples.
To use the cross-validation database interface, use the following:
from bob.bio.base.database import CSVDatasetCrossValidation
database = CSVDatasetCrossValidation(
name="my_cross_validation_dataset",
protocol="Default",
csv_file_name="your_dataset_name.csv",
test_size=0.8,
samples_for_enrollment=1,
csv_to_sample_loader=CSVToSampleLoader(
data_loader=bob.io.base.load,
dataset_original_directory="",
extension="",
metadata_loader=AnnotationsLoader()
),
)
The database interface class¶
Although most of the experiments will be satisfied with the CSV or cross-validation interfaces, there exists a way to specify exactly what is sent to each sub-pipelines by implementing your own database interface class.
When a PipelineSimple requests data from that class, it will call the following methods, which need to be implemented for each dataset:
bob.bio.base.pipelines.Database.background_model_samples(): Provides a list ofbob.pipelines.Sampleobjects that are used for training of the Transformers. Eachbob.pipelines.Samplemust contain at least the attributesbob.pipelines.Sample.keyandbob.pipelines.Sample.subject, as well as thebob.pipelines.Sample.dataof the sample.
bob.bio.base.pipelines.Database.references(): Provides a list ofbob.pipelines.SampleSetthat are used for enrollment of the models. The group (dev or eval) can be given as parameter to specify which set must be used. Eachbob.pipelines.SampleSetmust contain abob.pipelines.SampleSet.subjectattribute and a list ofbob.pipelines.Samplecontaining at least thebob.pipelines.Sample.keyattribute as well as thebob.pipelines.Sample.dataof the sample.
bob.bio.base.pipelines.Database.probes(): Returns a list ofbob.pipelines.SampleSetthat are used for scoring against a previously enrolled model. The group parameter (dev or eval) can be given to specify from which set of individuals the data comes. Eachbob.pipelines.SampleSetmust contain abob.pipelines.SampleSet.subject, abob.pipelines.SampleSet.referenceslist, and a list ofbob.pipelines.Samplecontaining at least thebob.pipelines.Sample.keyattribute as well as thebob.pipelines.Sample.dataof the sample.
Furthermore, the bob.bio.base.pipelines.Database.all_samples() method must return a list of all the existing samples in the dataset. This functionality is used for annotating a whole dataset.
Here is a code snippet of a simple database interface:
from bob.pipelines import Sample, SampleSet
from bob.bio.base.pipelines import Database
class CustomDatabase(Database):
def background_model_samples(self):
train_samples = []
for a_sample in dataset_train_subjects:
train_samples.append( Sample(data=a_sample.data, key=a_sample.sample_id) )
return train_samples
def references(self, group="dev"):
all_references = []
for a_subject in dataset_dev_subjects:
current_sampleset = SampleSet(samples=[], reference_id=a_subject.id)
for a_sample in a_subject:
current_sampleset.insert(-1, Sample(data=a_sample.data, key=a_sample.sample_id))
all_references.append(current_sampleset)
return all_references
def probes(self, group="dev"):
all_probes = []
for a_subject in dataset_dev_subjects:
current_sampleset = SampleSet(samples=[], reference_id=a_subject.id, references=list_of_references_id)
for a_sample in a_subject:
current_sampleset.insert(-1, Sample(data=a_sample.data, key=a_sample.sample_id))
all_probes.append(current_sampleset)
return all_probes
def all_samples(self, group=None):
all_subjects = dataset_train_subjects + dataset_dev_subjects
all_samples = []
for a_sample in all_subjects:
all_samples.append( Sample(data=a_sample.data, key=a_sample.sample_id) )
return all_samples
def groups(self):
return list(("train", "dev", "eval"))
def protocols(self):
return list(("protocol_1", "protocol_2"))
database = CustomDatabase()
Note
For optimization reasons, an score_all_vs_all
flag can be set in the database interface (see
bob.bio.base.pipelines.Database) to allow scoring
with all biometric references. This will be much faster when your algorithm
allows vectorization of operations, but not all protocols allow such a
feature.
When this flag is
True, the algorithm will compare a probe sample and generate the scores against every model of the set returned by thereferencesmethod.Otherwise (flag is
False), the scoring algorithm will only compare a probe to the givenbob.pipelines.SampleSet.references` attribute of its :py:class:`bob.pipelines.SampleSet.
Delayed samples¶
To work with datasets too big to fit entirely in memory, the bob.pipelines.DelayedSample was introduced.
The functionality is the same as a bob.pipelines.Sample, but instead
of storing the data as an attribute directly, a load funciton is used that
can be set to any function loading one sample of data. When data is needed, the
load function is called and the sample data is returned.
Checkpointing experiments¶
Checkpoints are a useful tool that allows an experiment to prevent computing data multiple times by saving the results of each step so it can be retrieved later. It can be used when an experiment fails in a later stage, preventing the computation of the stages coming before it, in case the experiment is restarted.
The checkpoints are files created on disk that contain the result of a sample of data passed through a Transformer or bob.bio.base.pipelines.BioAlgorithm.
When running, if the system finds a checkpoint file for its current processing step, it will load the results directly from the disk, instead of computing it again.
To enable the checkpointing of a Transformer or bob.bio.base.pipelines.BioAlgorithm, a bob.pipelines.CheckpointWrapper is available.
This class takes a Transformer as input and returns the same Transformer with the ability to automatically create checkpoint files.
The bob.pipelines.CheckpointWrapper class is available in the bob.pipelines.
The --checkpoint option is a command-line option that automatically wraps every steps of the pipeline with checkpointing.
If set, the --checkpoint-dir sets the path for such a checkpoints:
$ bob bio pipeline simple <database> <pipeline> --checkpoint --output <output_dir> --checkpoint-dir <checkpoint_dir>
When doing so, the output of each Transformer of the pipeline will be saved to the disk in the <checkpoint_dir> folder specified with the --checkpoint-dir option.
Output scores will be saved on <output_dir>.
Warning
You have to be careful when using checkpoints: If you modify an early step of an experiment, the created checkpoints are not valid anymore, but the system has no way of knowing that.
You have to take care of removing invalid checkpoints files.
When changing the pipeline or the dataset of an experiment, you should change
the output folder (--checkpoint-dir) accordingly. Otherwise, the system could try to
load a checkpoint of an older experiment, or samples from another dataset.
Scaling up with Dask¶
Dask is a library that allows advanced parallelism of python programs. This library takes care of creating jobs that can execute python code in parallel, schedules the execution of those jobs on the available resources, and manages the communication between those jobs. Dask jobs can run in parallel on the same machine, or be spread on a grid infrastructure. Diagnostic and monitoring tools are also available to watch progress and debug.
Using Dask with PipelineSimple¶
To run an experiment with Dask, a bob.pipelines.DaskWrapper class is available that takes any Transformer or bob.bio.base.pipelines.BioAlgorithm and outputs a dasked version of it.
You can easily benefit from Dask by using the --dask-client option like so:
$ bob bio pipeline simple <database> <pipeline> --dask-client <client-config>
where <client-config> is your own dask client configuration file, or a resource from bob.pipelines.distributed:
local-parallel: Launches as many jobs asmultiprocessing.cpu_count()that will run simultaneously on the current machine.sge: Runs jobs on a grid architecture, on any workers (at Idiap, uses the SGE. Other grids may require more configuration, seebob.pipelines).sge-gpu: Executes jobs on a grid architecture, but defaults to the nodes with a GPU (q_short_gpu).
Note
If the dask option is not set (omitting the -l), everything will run locally in one single thread.
Warning
For Idiap users: If you need to run the PipelineSimple in the SGE. Don’t forget to do:
$ SETSHELL grid
Also, since the grid nodes are not allowed to create additional jobs on the grid, you cannot run the main dask client on a job (qsub -- bob bio pipeline simple -l sge_... will not work).
Monitoring and diagnostic¶
A running instance of Dask will create a server with a listening port (default: port 8787) that answers to HTTP requests. This means that when an instance of Dask is running, you can open localhost:8787 in your browser to monitor the running jobs.
The page available contains some useful tools like a graphical visualization of the jobs and their dependencies, a resources monitor (CPU and memory of each node), and access to the logs of all running and finished jobs.
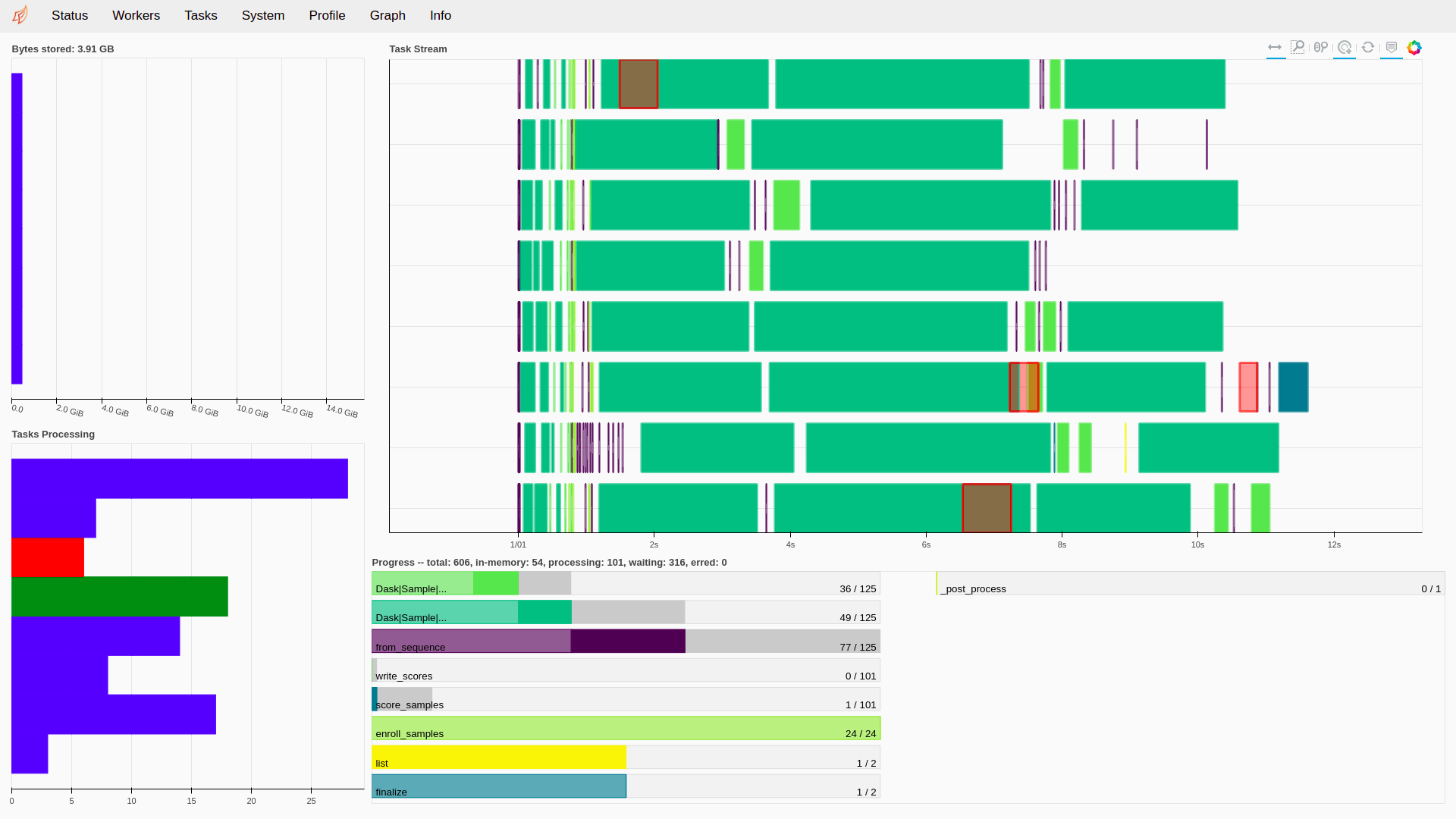
Fig. 9 The Status page of the Dask monitoring tool. 8 jobs are running in parallel with different steps being processed.¶
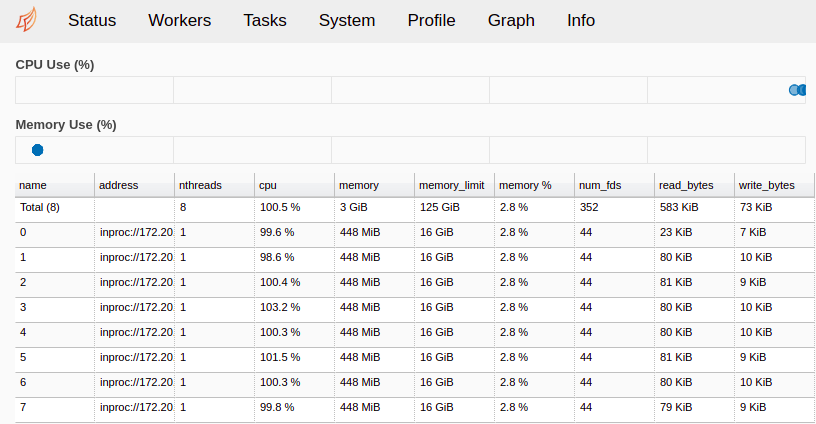
Fig. 10 The Workers page of the Dask monitoring tool. 8 workers are accepting jobs, and their current memory and CPU usage are plotted.¶
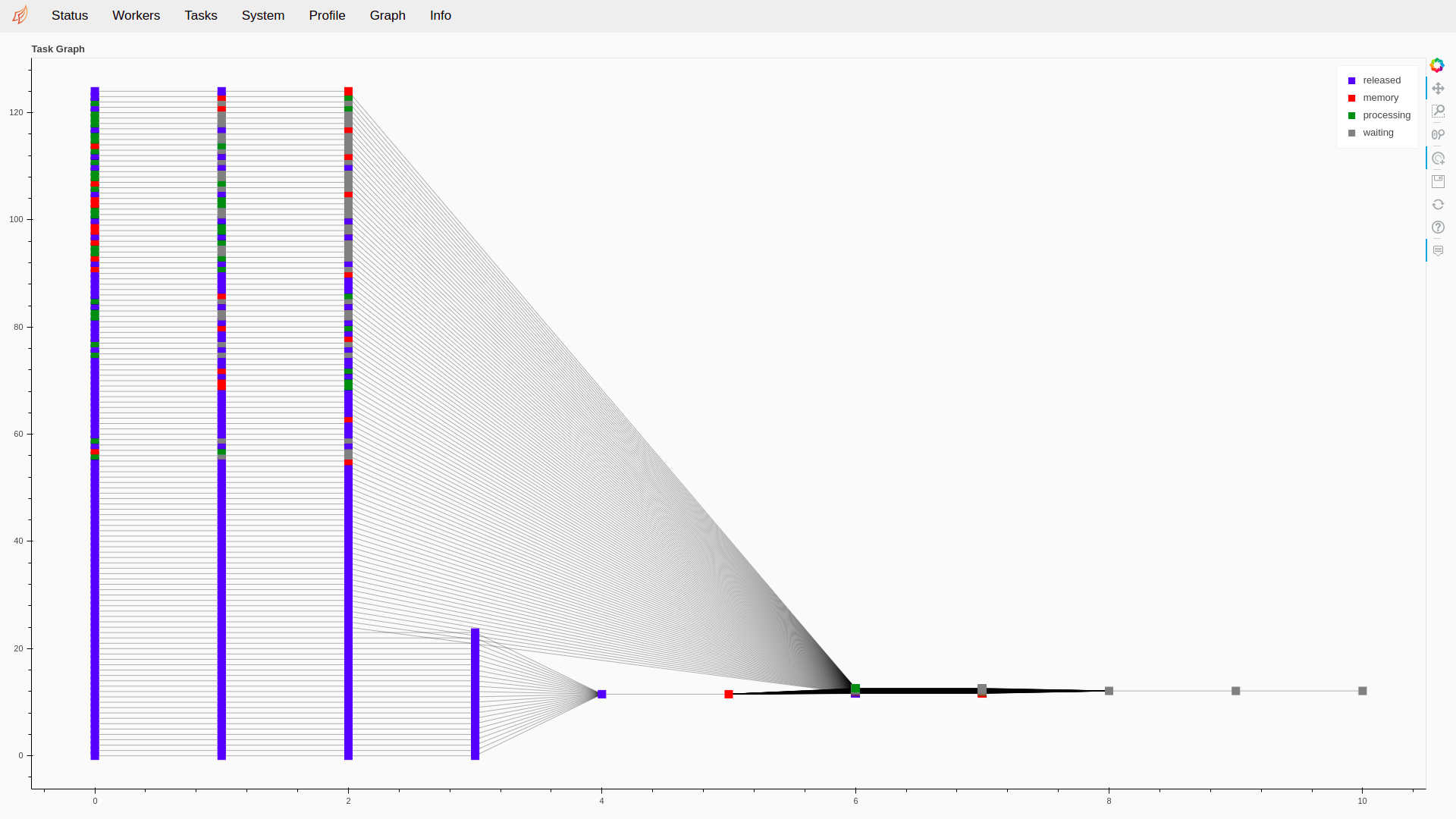
Fig. 11 The Graph page of the Dask monitoring tool. We can observe the different steps of a biometric experiment, with the bottom part being the enrollment of references and the top part the probes used for scoring. The input are on the left side, and the resulting scores are generated by the jobs on the right.¶
Writing scores in a customized manner¶
The results of the PipelineSimple are scores that can be analyzed.
To do so, the best way is to store them on disk so that a utility like bob bio metrics or bob bio roc can retrieve them.
However, many formats could be used to store those score files, so bob.bio.base.pipelines.ScoreWriter was defined.
A bob.bio.base.pipelines.ScoreWriter must implement a bob.bio.base.pipelines.ScoreWriter.write() method that receives a list of bob.pipelines.SampleSet that contains scores as well as metadata (subject identity, sample path, etc.).
Common bob.bio.base.pipelines.ScoreWriter are available by default:
A CSV format ScoreWriter
A four columns format ScoreWriter
Using a specific Score Writer¶
By default, PipelineSimple will use the CSV format ScoreWriter.
To indicate to a pipeline to use the four-columns ScoreWriter instead
of the default CSV ScoreWriter, you can pass the --write-column-scores option like so:
$ bob bio pipeline simple --write-column-scores <database> <pipeline> --output <output_dir>
CSV Score Writer¶
A bob.bio.base.pipelines.CSVScoreWriter is available, creating a Comma Separated Values (CSV) file with all the available metadata of the dataset as well as the score of each comparison.
It is more complete than the bob.bio.base.pipelines.FourColumnsScoreWriter and allows analysis of scores according to sample metadata (useful to analyze bias due to e.g. age or gender).
The default bob.bio.base.pipelines.CSVScoreWriter will write a probe subject and key, a biometric reference subject, the score resulting from the comparison of that probe against the reference, as well as all the fields set in bob.pipelines.SampleSet by the database interface, for the references and the probes.
A header is present, to identify the metadata, and the values are comma separated.
Here is a short example of such file format with metadata on the age and gender of the subject:
probe_subject, probe_key, probe_age, probe_gender, bio_ref_subject, bio_ref_age, bio_ref_gender, score
subj_0, subj0_sample0, 27, male, subj_0, 29, male, 0.966
subj_0, subj0_sample0, 29, male, subj_1, 45, female, 0.127
subj_1, subj1_sample0, 45, female, subj_2, 40, female, 0.254
subj_1, subj1_sample1, 46, female, subj_2, 40, female, 0.287
Four Columns Score Writer¶
bob.bio.base.pipelines.FourColumnsScoreWriter is the old score file format used by bob.
It consists of a text file with four columns separated by spaces and no header.
Each row represents a comparison between a probe and a model, and the similarity score resulting from that comparison.
The four columns are, in order:
Reference ID: The identity of the reference or model used for that comparison (field
subjectinbob.pipelines.SampleSetof thereferencesset).Probe ID: The identity of the probe used for that comparison (field
subjectinbob.pipelines.SampleSetof theprobesset).Probe key: A unique identifier (often a file path) for the sample used as probe for that comparison.
Score: The similarity score of that comparison.
Here is a short example of such file format:
subj_0 subj_0 subj0_sample0 0.998
subj_0 subj_0 subj0_sample1 0.985
subj_0 subj_1 subj1_sample0 0.450
subj_0 subj_1 subj1_sample1 0.289
Evaluation¶
Once scores are generated for each probe evaluated against the corresponding models, conclusions on the performance of the system can be drawn by using metrics like the FMR and FNMR or tools like the ROC curve.
In this section, commands that help to quickly evaluate a set of scores by generating metrics or plots are presented. The scripts take as input either a csv file or a legacy four/five column format. See Writing scores in a customized manner for the explanation of their formats.
Note
If both dev and eval scores are available, the scores of the dev set will be used to define the threshold used to evaluate the scores of the eval set.
Metrics¶
To calculate the threshold using a certain criterion (EER (default), FAR, or min.HTER) on a development set and apply it on an evaluation set, just do:
$ bob bio metrics -e output/scores-{dev,eval}.csv --legends ExpA --criterion min-hter
[Min. criterion: MIN-HTER ] Threshold on Development set `ExpA`: -1.399200e-01
===================== ============== ==============
.. Development Evaluation
===================== ============== ==============
Failure to Acquire 0.0% 0.0%
False Match Rate 2.4% (24/1000) 1.9% (19/1000)
False Non Match Rate 0.0% (0/250) 2.0% (5/250)
False Accept Rate 2.4% 1.9%
False Reject Rate 0.0% 2.0%
Half Total Error Rate 1.2% 1.9%
===================== ============== ==============
Note
When evaluation scores are provided, the --eval option must be passed.
See metrics --help for further options.
The scores-{dev,eval}.csv brace expansion (a bash feature) expands the
path to every element in the list. here: scores-dev.csv scores-eval.csv.
Plots¶
Customizable plotting sub-commands are available in the bob.bio.base
module (command: $ bob bio). They take a list of development and/or
evaluation files and generate a single PDF file containing the plots.
Available plots are:
roc(receiver operating characteristic)det(detection error trade-off)epc(expected performance curve)hist(histograms of scores with threshold line)cmc(cumulative match characteristic)dir(detection & identification rate)
Use the --help option on the above-cited commands to find-out about more
options.
For example, to generate a CMC curve from development and evaluation datasets:
$ bob bio cmc -v --eval --output 'my_cmc.pdf' dev-1.csv eval-1.csv dev-2.csv eval-2.csv
where my_cmc.pdf will contain CMC curves for the two experiments represented
by their respective dev and eval scores-file.
Note
By default, det, roc, cmc, and dir plot development and
evaluation curves on different plots. You can forcefully gather everything in
the same plot using the --no-split option.
Note
The --figsize and --style options are two powerful options that can
dramatically change the appearance of your figures. Try them! (e.g.
--figsize 12,10 --style grayscale)
Bob bio evaluate¶
A convenient command evaluate is provided to generate multiple metrics and
plots for a list of experiments. It generates two metrics outputs with EER,
HTER, minDCF criteria, along with roc, det, epc, and hist plots
for each experiment. For example:
$ bob bio evaluate -v --eval --log 'my_metrics.txt' --output 'my_plots.pdf' {sys1,sys2}/scores-{dev,eval}.csv
will output metrics and plots for the two experiments (dev and eval pairs) in
my_metrics.txt and my_plots.pdf, respectively.
Transforming samples¶
It is possible to use the a “part” of the PipelineSimple to transform the samples in a given dataset. This is useful for:
Crop all faces from a dataset
Extract features using a particular feature extractor
Preprocess some audio files using a particular pre-processor
This can be done with the command:
bob bio pipeline transform --help
Options:
-t, --transformer CUSTOM A scikit-learn Pipeline containing the set of
transformations Can be a
``bob.bio.transformer`` entry point, a module
name, or a path to a Python file which contains
a variable named `transformer`. [required]
-d, --database CUSTOM Biometric Database connector (class that
implements the methods:
`background_model_samples`, `references` and
`probes`) Can be a ``bob.bio.database`` entry
point, a module name, or a path to a Python
file which contains a variable named
`database`. [required]
-l, --dask-client TEXT Dask client for the execution of the pipeline.
Can be a ``dask.client`` entry point, a module
name, or a path to a Python file which contains
a variable named `dask_client`.
-c, --checkpoint-dir TEXT Name of output directory where the checkpoints
will be saved. [default: ./checkpoints]
-e, --file-extension CUSTOM File extension of the output files. [required]
-f, --force If set, it will force generate all the
checkpoints of an experiment. This option
doesn't work if `--memory` is set
-v, --verbose Increase the verbosity level from 0 (only error
messages) to 1 (warnings), 2 (log messages), 3
(debug information) by adding the --verbose
option as often as desired (e.g. '-vvv' for
debug).
-H, --dump-config FILENAME Name of the config file to be generated
-?, -h, --help Show this message and exit.
Warning
It is assumed that the pipeline is Sample wrapped.
The option –transformer accepts a python resource as an argument, but also can accept a regular Python file which contains a variable named transformer defining the Pipeline like the example below:
from sklearn.base import BaseEstimator, TransformerMixin
from sklearn.pipeline import make_pipeline
from bob.pipelines import wrap
class MyTransformer(TransformerMixin, BaseEstimator):
def _more_tags(self):
return {"requires_fit": False}
def transform(self, X):
# do something
return X
transformer = wrap(["sample"], make_pipeline(MyTransformer()))
Then saving this to a file called my_beautiful_transformer.py and passing it as an argument bob bio pipelines transform command:
$ bob bio pipelines transform my_database my_beautiful_transformer.py
Note
It is possible to leverage from Dask by setting the option –dask-client the same way it is done with the bob bio pipeline simple command
If you are skilled on scikit learn, it is possible to leverage from FunctionTransformer function and make the above Transformer definition way simpler as in the example below:
from sklearn.preprocessing import FunctionTransformer
from sklearn.pipeline import make_pipeline
from bob.pipelines import wrap
def my_transformer(X):
# do something
return X
transformer = wrap(["sample"], make_pipeline(FunctionTransformer(my_transformer)))
It is also possible to provide a pipeline that is already checkpointed. For instance, this is useful when you and to change the way a pipeline saves a particular transformed output or if you want to checkpoint only one part of a pipeline (e.g., you need to save only the features and not preprocessed samples).
from sklearn.preprocessing import FunctionTransformer
from sklearn.pipeline import make_pipeline
from bob.pipelines import wrap
def my_transformer(X):
# do something
return X
transformer = wrap(["sample"], make_pipeline(FunctionTransformer(my_transformer)))
transformer = wrap(["checkpoint"], transformer,
features_dir=MY_DIR,
extension=MY_EXTENSION,
save_func=SAVE_FUNCTION_YOU_WANT_TO_USE),
)
Warning
The order of the transformation matter. For instance, for face recognition experiments to get features properly extracted, you should run the command as:
$ bob bio pipelines transform my_database my_beautiful_transformer.py