Vanilla Biometrics: Introduction to biometric recognition in practice¶
Note
Make sure the following conda packages are installed before following this tutorial:
conda install bob.bio.base bob.bio.face bob.db.atnt
Also note that almost all bob.db.* packages are deprecated and they should
not be installed. The bob.bio.face package contains the implementation of
all face biometric databases. Same is true for other bob.bio packages.
To run biometric experiments, we provide a generic CLI command called bob bio pipelines.
Such CLI command is an entry-point to several pipelines implemented in this package.
Curently only one pipeline is implemented which is vanilla-biometrics.
This tutorial will focus on this pipeline.
In our very first example, we’ve shown how to compare two samples using the
bob bio compare-samples command, where the “biometric” algorithm is set with
the argument --pipeline. A pipeline is an instance of
bob.bio.base.pipelines.vanilla_biometrics.VanillaBiometricsPipeline.
Running a biometric experiment with Vanilla Biometrics¶
A bob bio pipelines vanilla-biometrics command is available to run Vanilla Biometrics experiments from the shell.
Its options can be listed with:
$ bob bio pipelines vanilla-biometrics --help
The command accepts a pipeline and a database to run the experiment.
Building your own Vanilla Biometrics pipeline¶
The Vanilla Biometrics represents the simplest biometrics pipeline possible and for this reason, is the backbone for any biometric test in this library. It’s composed of:
One or several Transformers: Instances of
sklearn.base.BaseEstimatorandsklearn.base.TransformerMixin. A Transformer can be trained if needed and applies one or several transformations on an input sample. It must implement aTransformer.transformand aTransformer.fit()method. Multiple transformers can be chained together, each working on the output of the previous one.A Biometric Algorithm: Instance of
bob.bio.base.pipelines.vanilla_biometrics.BioAlgorithmthat implements the methodsenrollandscoreto generate biometric experiment results.
Running the vanilla-biometric pipeline will retrieve samples from a dataset and generate score files.
It does not encompass the analysis of those scores (Error rates, ROC, DET). This can be done with other utilities of the bob.bio packages.
Transformer¶
Following the structure of
pipelines of scikit-learn,
a Transformer is a class that must implement a Transformer.transform
and a Transformer.fit() method. This class represents a simple
operation that can be applied to data, like preprocessing of a sample or
extraction of a feature vector from data.
A Transformer must implement the following methods:
- Transformer.transform(X)¶
This method takes data (
X) as input and returns the corresponding transformed data. It is used for preprocessing and extraction.
- Transformer.fit(X, y=None)¶
A Transformer can be trained with its
Transformer.fit()method. For example, for Linear Discriminant Analysis (LDA), the algorithm must first be trained on data.This method returns the instance of the class itself (
self).
Note
Not all Transformers need to be trained (via a fit method).
For example, a preprocessing step that crops an image to a certain size does not require training. In this case, the Transformer.fit() method simply returns self.
It is best to use sklearn.preprocessing.FunctionTransformer to create a transformer that does not require fit.
Below is an example implementing a very simple Transformer applying a custom function on each sample given as input.
from sklearn.base import TransformerMixin, BaseEstimator
class CustomTransformer(TransformerMixin, BaseEstimator):
def transform(self, X):
transformed_X = my_function(X)
return transformed_X
def fit(self, X, y=None):
return self
or using sklearn.preprocessing.FunctionTransformer:
from sklearn.preprocessing import FunctionTransformer
def CustomTransformer(**kwargs):
return FunctionTransformer(my_function, **kwargs)
Biometric Algorithm¶
A biometric algorithm represents the enrollment and scoring phase of a biometric experiment.
A biometric algorithm is a class implementing the method
bob.bio.base.pipelines.vanilla_biometrics.BioAlgorithm.enroll() that
allows to save the identity representation of a subject, and
bob.bio.base.pipelines.vanilla_biometrics.BioAlgorithm.score()
that computes the score of a subject’s sample against a previously enrolled
model.
A common example of a biometric algorithm class would compute the mean vector of the features of each enrolled subject, and the scoring would be done by measuring the distance between the unknown identity vector and the enrolled mean vector.
- BiometricAlgorithm.enroll(reference_sample)¶
The
bob.bio.base.pipelines.vanilla_biometrics.BioAlgorithm.enroll()method takes extracted features (data that went through transformers) of the reference samples as input. It should save (on memory or disk) a representation of the identity of each subject for later comparison with thebob.bio.base.pipelines.vanilla_biometrics.BioAlgorithm.score()method.
- BiometricAlgorithm.score(model, probe_sample)¶
The
bob.bio.base.pipelines.vanilla_biometrics.BioAlgorithm.scoremethod also takes extracted features (data that went through transformers) as input but coming from the probe samples. It should compare the probe sample to the model and output a similarity score.
Here is a simple example of a custom bob.bio.base.pipelines.vanilla_biometrics.BioAlgorithm implementation that computes a model with the mean of multiple reference samples, and measures the inverse of the distance as a similarity score.
from bob.bio.base.pipelines.vanilla_biometrics import BioAlgorithm
class CustomDistance(BioAlgorithm):
def enroll(self, enroll_features):
model = numpy.mean(enroll_features, axis=0)
return model
def score(self, model, probe):
distance = 1/numpy.linalg.norm(model-probe)
return distance
Constructing the pipeline¶
As stated before, a pipeline is a series of Transformers and a BiometricAlgorithm chained together. In Vanilla biometrics, 3 sub-pipelines are defined: a training pipeline, an enrollment pipeline, and a scoring pipeline.
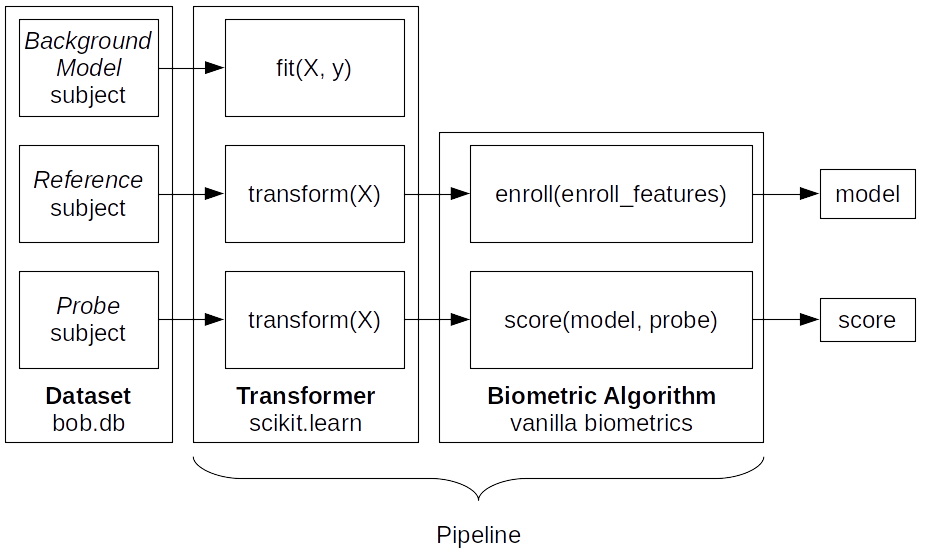
Fig. 8 Example of a pipeline showing the sub-pipelines. The data of references is used for enrollment and the data of probes is used for scoring.
Each subject’s data goes through the Transformer (or series of Transformers) before being given to BiometricAlgorithm.enroll() or BiometricAlgorithm.score().¶
Here is the creation of the pipeline combining the Transformer and the BioAlgorithm that we implemented earlier:
from sklearn.pipeline import make_pipeline
from bob.pipelines import wrap
from bob.bio.base.pipelines.vanilla_biometrics import VanillaBiometricsPipeline
# Instantiate the Transformer(s)
my_transformer = CustomTransformer()
# make it a sample transformer (explained later)
my_transformer = wrap(["sample"], my_transformer)
# Chain the Transformers together
transformer = make_pipeline(
my_transformer,
# Add more transformers here if needed
)
# Instantiate the BioAlgorithm
bio_algorithm = CustomDistance()
# Assemble the Vanilla Biometric pipeline and execute
pipeline = VanillaBiometricsPipeline(transformer, bio_algorithm)
Minimal example of a vanilla-biometrics experiment¶
To run a minimal example, let’s download the ATNT faces database and execute this pipeline. The ATNT database can be easily downloaded using the following command:
$ bob_dbmanage.py atnt download --output-dir ~/bob_data/datasets/atnt
Note
Usually, you need to download the files of each database manually yourself. We do not and cannot provide a script that downloads a biometric database automatically.
For each database, you need to configure Bob to specify the location of its files. To do so for ATNT, run the following command:
$ bob config set bob.db.atnt.directory ~/bob_data/datasets/atnt
For more information, see Global Configuration System.
Find below a complete file containing a Transformer, a Biometric Algorithm, and the construction of the pipeline:
import numpy as np
from bob.bio.base.pipelines.vanilla_biometrics import BioAlgorithm
from bob.bio.base.pipelines.vanilla_biometrics import VanillaBiometricsPipeline
from bob.pipelines import wrap
from sklearn.decomposition import PCA
from sklearn.pipeline import make_pipeline
from sklearn.preprocessing import FunctionTransformer
from sklearn.utils import check_array
## Transformers
pca = PCA(n_components=0.95)
# the images are in shape of Nx112x92, we want to flatten to Nx10304 them so we can train a PCA on them.
# A similar implementation is available in:
# from bob.pipelines.transformers import Linearize
def flatten(images):
images = check_array(images, allow_nd=True)
new_shape = [images.shape[0], -1]
return np.reshape(images, new_shape)
flatten_transformer = FunctionTransformer(flatten, validate=False)
# Chain the Transformers together
transformer = make_pipeline(flatten_transformer, pca)
# All transformers must be sample transformers
transformer = wrap(["sample"], transformer)
## Implementation of the BioAlgorithm
# A better implementation is available in:
# from bob.bio.base.pipelines.vanilla_biometrics import Distance
class EuclideanDistance(BioAlgorithm):
def enroll(self, enroll_features):
model = np.mean(enroll_features, axis=0)
return model
def score(self, model, probe):
similarity = 1/np.linalg.norm(model-probe)
# you should always return a similarity score
return similarity
bio_algorithm = EuclideanDistance()
## Creation of the pipeline
# `pipeline` will be used by the `bob bio pipelines vanilla-biometrics` command
pipeline = VanillaBiometricsPipeline(transformer, bio_algorithm)
# you can also specify the other options in this file:
database = "atnt"
output = "results"
To run the simple example above, save that code in a file my_pipeline.py and enter this command in a terminal:
$ bob bio pipelines vanilla-biometrics /path/to/my_pipeline.py
Note
You can specify all options in one .py file above when
providing the config file as an argument, like the example.
To create a sample config file, run:
$ bob bio pipelines vanilla-biometrics -H sample_config.py
This will create a file results/scores-dev containing the distance between each pair of probe and reference sample.
Structure of a pipeline¶
In a serious scenario with more complex and longer implementations, you should separate the definition of Transformers and BioAlgorithm in different files that can be swapped more easily.
bob.bio packages also provide commonly used pipelines and databases that you can use. You can list them with the following command:
$ resources.py
For example, to test the gabor graph pipeline on the ATNT database, run:
$ bob bio pipelines vanilla-biometrics -vv atnt gabor_graph
The command above is equivalent to the following command:
$ bob bio pipelines vanilla-biometrics -vv \
bob.bio.face.config.database.atnt \
bob.bio.face.config.baseline.gabor_graph
This information can obtained using resources.py:
$ resources.py --type config
+ atnt --> bob.bio.face.config.database.atnt
+ gabor_graph --> bob.bio.face.config.baseline.gabor_graph
See Extending packages as frameworks for more information.
Note
Many pipelines depend on the fact that you run them like:
bob bio pipelines vanilla-biometrics -vv <database> <pipeline>
where no --database and --pipeline is used and the database
is specified before the pipeline.